 |
||
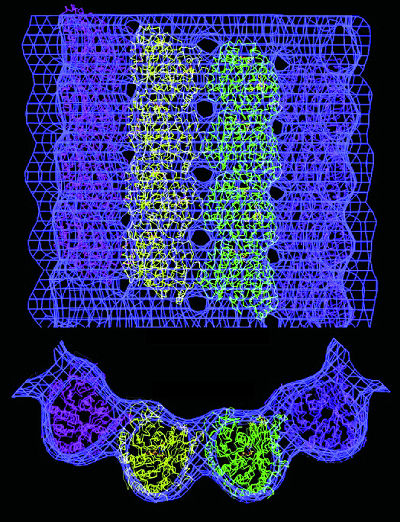
| Electron microscopy can provide 3-D atomic-scale models of proteins that are difficult to image with x-rays. This model shows a microtuble, a hollow cylindrical structure made up of tubulin proteins that serves as part of the skeletal system for cells and is crucial to a number of vital functions including mitosis. Tubulin is the protein that interacts with the anti-cancer drug taxol. | ||
Seeing the Bigger Picture
The beam of an electron microscope can produce diffraction patterns from two-dimensional crystalline arrays rather than the three-dimensional arrays required for x-ray diffraction. In the case of membrane proteins, these 2-D crystals can be created in a more natural setting-sheets of fatty molecules called lipids-than the heavy concentrations of detergents needed to form 3-D crystals.
Sometimes electron crystallography is the only ![]() way
to go, as was the case two years ago when biophysicists Eva Nogales, Sharon
Wolf, and Kenneth Downing, of Berkeley Lab's Life Sciences Division, announced
the completion of the first 3-dimensional atomic model of a protein called
"tubulin," a highly versatile molecule which, among other functions, enables
a cell to undergo mitosis. At a resolution of 3.7 angstroms, this model
provided the first highly-detailed three-dimensional look at tubulin,
including the site where the protein interacts with the anti-cancer drug
taxol.
way
to go, as was the case two years ago when biophysicists Eva Nogales, Sharon
Wolf, and Kenneth Downing, of Berkeley Lab's Life Sciences Division, announced
the completion of the first 3-dimensional atomic model of a protein called
"tubulin," a highly versatile molecule which, among other functions, enables
a cell to undergo mitosis. At a resolution of 3.7 angstroms, this model
provided the first highly-detailed three-dimensional look at tubulin,
including the site where the protein interacts with the anti-cancer drug
taxol.
The use of electron-based rather than x-ray-based crystallography techniques was crucial to the creation of this model. As Downing explained, "obtaining diffraction patterns with an electron beam enabled us to work with crystals only one molecule in thickness, giving us our high resolution."
Solving tubulin's atomic structure was a spectacular success-scientists had been trying to do it since the protein's discovery in the 1960s-but the project took the Berkeley Lab researchers more than six years to complete. During that time, they recorded some 4,000 images and electron diffraction patterns which Nogales processed to come up with the 159 images and 93 diffraction patterns used to do a computer reconstruction for the final 3-D tubulin model.
The technique used to solve the tubulin structure is called cryo-electron microscopy or "cryo-em" because the images were recorded with an electron microscope equipped with a "cold stage." Freezing the protein preserves it in its native hydrated state and helps protect it from radiation damage. Another addition to electron microscopy with exceptionally strong potential for solving the structures of macromolecular protein complexes is a technique called "single-particle image analysis."
 |
|
| Kenneth Downing (left), Eva Nogales and Robert Glaeser have been experimenting with "crystallization in silico," the use of supercomputers to construct 3-D models based on millions of electron microscopy images of single non-crystallized proteins. | |
In single-particle image analysis, thousands of images of randomly-oriented individual protein molecules are recorded via an electron microscope. A computer is then used to align these thousands of randomly-oriented images into an ordered array and merge them into a three-dimensional reconstruction-in essence, creating a virtual crystal.
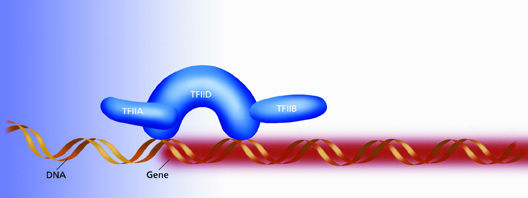
The power of single-particle image analyis as a tool for structural genomics was recently demonstrated in a project led by Nogales. Working with proteins prepared in the lab of UC Berkeley professor Robert Tjian, Nogales and Frank Andel used the combination of electron microscopy and single particle image analysis to produce the first 3-D models of the protein machinery that initiates the transcription of the genetic code of dna for the subsequent production of new proteins. This machinery is actually a complex of transcriptional factor (tf) proteins that include TFIID, TFIIA and TFIIB.
After a strand of dna has been unwound and unzipped in preparation for protein production, the TFIID protein binds to an exposed dna section precisely where a genetic message begins. Once TFIID recognizes and binds to a gene on a strand of dna, it interacts with rna polymerase so that the genetic code is transcribed into messanger rna, which then carries the information out of the cell's nucleus and into its cytoplasm where protein assembly takes place.
"Our model gives us a good idea as to how TFIID works in concert with TFIIA and TFIIB to initiate and regulate the transcription of protein coding genes," says Nogales. "We show a horseshoe-shaped structure surrounding a central cavity inside of which recognition and binding to dna takes place."
Structural determinations for some domains and other components of the
TFIID, TFIIA and TFIIB proteins have been determined through x-ray crystallography
but the size of TFIID, coupled with the difficulties posed in trying to
crystallize all of the tf proteins together, has so far precluded the
use of x-ray diffraction for imaging the entire complex. Consequently,
until now the overall shape and relative position of the components within
the complex were a mystery.
 |
|
| TFIID is a horseshoe-shaped protein that starts the process of gene transcription by binding to an exposed strand of DNA precisely where a genetic message begins. The central cavity of the horseshow is sized to easily fasten around a single strand of DNA. | |
Because Nogales and her colleagues did not have to crystallize the tf complex, they could work with a relatively small amount of sample and still produce an image of the entire transcriptional machine. In less than seven months, they had their 3-D reconstruction at a resolution of 35 angstroms.
"Our study shows that electron microscopy is a good technique for studying biological complexes of proteins and nucleic acids that are too large or too fragile to be crystallized for x-ray diffraction studies," says Nogales, who credits Andel for the single-particle analysis work. "Our study also shows that single-particle methodology is a useful technique for the structural characterization of mega-Dalton transcription complexes."
At a resolution of 35 angstroms the shapes of TFIID and its companion proteins plus their relative positions within the tf complex can be clearly seen. When this electron microscopy information is combined with the x-ray data on various substructures within the complex-a technique dubbed "hybrid crystallography"-Nogales and her colleagues expect to find further clues as to how the transcriptional machinery comes together. (More about the Nogales lab at http://cryoem.berkeley.edu.)
Robert Glaeser, a professor of biophysics at UC Berkeley with a joint appointment in Berkeley Lab's Life Sciences and Physical Biosciences Divisions, is a pioneer and world authority on electron crystallography. Glaser says that hybrid crystallography is raising "a great amount of enthusiasm" among the scientists in his field because "it is the complexes rather than the individual components that really begin to bring structural biology into the realm of functional genomics."
Glaeser compares x-ray crystallography with identifying all the individual components that go into an auto engine block-spark plugs, cam shaft, valves, etc. "The structure of each of these components is very interesting and enlightening but you also want to know how the spark plug fits into the block, the order in which the valves open and close, and so forth, and how all of that is accomplished," he says.
Glaeser, Nogales, and Downing have recently begun using an electron microscope at Berkeley Lab's National Center for Electron Microscopy (ncem) that is equipped with a field emission gun which enables it to direct highly focused electron beams at 200 kilovolts (kVs) of energy onto a sample. This sharpens the resolution and boosts the contrast to yield better quality images than the lower-powered electron microscopes they'd previously used. The three are overseeing the acquisition of new custom-built electron microscope that will accelerate electron beams to 300 kV which should improve resolution and image quality even further.
"This new microscope will allow us to get the maximum resolution possible in our images while doing the least amount of damage to our samples," says Nogales. "With the addition of new detector technology, we will also be able to collect our data much faster and more efficiently."
Between the new microscope and the facilities at ncem, the Berkeley Lab researchers hope to be able to record up to 3,000 images a day. Still, electron microscopy and hybrid crystallography won't match synchrotron-based x-ray crystallography for atomic-level details and rate of throughput. Another approach to electron microscopy, however, might come close.
Glaeser, in collaboration with Downing and Nogales, plus Ravi Malladi and Esmond Ng of the National Energy Research Scientific Computing Center (nersc), is exploring the viability of applying the enormous computational powers of a supercomputer to electron microscopy imaging techniques.
With a supercomputer, rather than working with a few tens of thousands of images as is now done, it should be possible to collect and merge as many as a million images of a single non-crystallized protein, then reconstruct these images into a 3-D model at a resolution comparable to that achieved with x-ray crystallography. What's more, with supercomputers at nersc capable of making trillions of calculations every second, such reconstructions could be done in less than half-a-day. Glaeser calls this approach "crystallization in silico," the equivalent of using a computer to perform the difficult task of protein crystallization.
 |
||
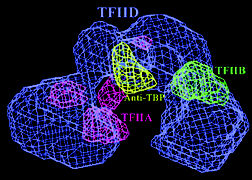
| A model reconstructed from electron microscopy images shows that TFIID's central cavity is defined by TFIIA and TFIIB. Antibody mapping of TBP, the protein that actually binds to DNA, located it at the top of the cavity. | ||
Glaeser and his collaborators aim to make crystallization in silico "an important player in structural genomics" by looking into ways of automating image data-collection, particle identification, and the final merging of data from individual particles.
"With further improvements in image quality, more closely approximating what the laws of physics will allow, the particle size that can be handled by this technique could push down to the range that includes all but the smallest proteins," Glaeser says.
"At that stage, it might become the method of choice for solving the
structures of any protein that has not already been crystallized."
Zooming in on the Details
With x-ray crystallography to determine the 3-D structures of protein components and electron microscopy to show how these components fit into complexes, there is still a need to zoom in for the finer details of electronic properties in order to see how a protein machine performs its chemical functions. The best techniques for this work are x-ray absorption spectroscopy (xas), and extended x-ray absorption fine structure (exafs). Both techniques are based upon the energy spectrum produced when one or more core electrons (those whose energy orbitals are closest to the nuclei of an atom) absorb x-rays. In xas, the electron is bumped up to a higher energy orbital and the resulting spectrum provides important information on how the host atom's electrons are configured. In exafs, the electron is ejected from the atom as a photoelectron and the resulting spectrum provides extremely accurate measurements of electron bond lengths.
"X-ray absorption spectroscopy is a wonderful complement to x-ray crystallography," says pbd chemist Karen McFarlane. "We can look at a specific type of atom within the protein and see who its neighbors are, and we can look at it under several different environments or after we've manipulated it."
McFarlane oversaw the design and construction of a new experimental endstation at als beamline 9.3.1, a bend magnet beamline that generates x-ray photons in the 2 to 6 thousand electron volt (keV) range. These photons are ideal for the study of atoms such as sulfur, chlorine, and calcium, which are minor but critically important protein constituents. For example, a sulfur-containing peptide, glutathione, helps prevent oxidative and free-radical damage to the cell, and strengthens red blood cells while protecting white blood cells.
 |
|
| Karen McFarlane and Mel Klein at the experimental endstation of ALS beamline 9.3.1 where XAS and EXAFS techniques can be used to study a protein's electronic properties. These properties yield valuable clues about the protein's chemical functions. | |
The new endstation at beamline 9.3.1 features a sample chamber with a liquid helium cryostat for cooling protein molecules down to 10 Kelvin in order to minimize radiation damage and allow exafs in addition to xas data collection. Pressure within the chamber can be varied or experiments can be done in vacuum, and samples can be studied in a solid or liquid state. This unique chamber was built by pbd physicist Mel Klein, who is the principal investigator for research at the endstation, working in collaboration with fellow pbd chemist Vittal Yachandra.
"Until now, most xas and exafs experiments have been done on the metal centers in metalloproteins while experiments with sulfur, chlorine, calcium and other lower-Z atoms have been rare," says McFarlane. "We plan to use our experimental station extensively for sulfur in many different proteins, peptides, and inorganic complexes. We also have plans for probing chlorine and calcium in the Photosystem II protein complex."
In addition to providing electronic and structural information that will complement x-ray crystallography data, x-ray spectroscopy techniques can also be used to perform comparison measurements that will answer questions neither technique could answer alone. For example, such comparison measurements could help determine whether there are differences in the active sites of proteins when they are in solution as opposed to when they have been crystallized. Experiments on the xas/exafs endstation at beamline 9.3.1 are scheduled to begin early next summer.
Making
Protein Movies
Whether focusing on the biggest, smallest, or finest details of protein structures, all of the techniques so far discussed have one thing in common-all provide static images. Yet protein machines are dynamic when they perform their functions. Just as a photograph of a car at rest, no matter how high the image quality, yields little appreciation for what the car will do when it moves, scientists will need to see a protein in action to fully understand its purpose.
New technologies now being developed make it possible to obtain dynamic images of proteins. Rather than providing composite images reconstructed from an ensemble or aggregate of a select type of protein molecule, these new technologies enable scientists to study and even physically manipulate a single individual molecule.
Shimon Weiss, a physicist who holds a joint appointment with the pbd and the Materials Sciences Division (msd), is one of the recognized leaders in single-molecule spectroscopy. He explains the advantage of characterizing and manipulating an individual protein molecule.
"In contrast with ensemble measurements, single-molecule experiments provide information on distributions and time trajectories that would otherwise be hidden. In particular, single-molecule experiments can be used to measure intermediates and follow time-dependent pathways of chemical reactions that are difficult or impossible to synchronize at the ensemble level."
 |
||
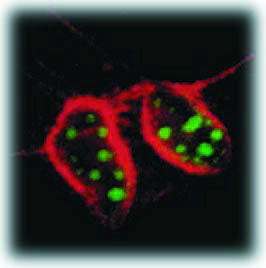
| In this cross-section of mouse cells labeled with two different sizes of fluorescing semiconductor nanocrystals, nuclei show up as green and actin fibers show up as red under the same illuminating beam. | ||
The general strategy behind single molecule spectroscopy is to attach dye molecules to various sites along a protein. These dye molecules fluoresce in a specific color when zapped by laser light. Tracking the intensity and location of fluorescent emissions over time reveals any changes that may be taking place in the protein's structure. A variation of this strategy that is especially valuable for measuring protein conformational changes is "fluorescence resonance energy transfer" or fret.
To use fret, scientists tag a protein with a pair of dye molecules, one acting as an energy "donor" and the other as an energy "acceptor." The fret effect is observed when the donor is photo-excited and its fluorescence energy matches the energy the acceptor will absorb. If the protein contracts so as to bring the two tags closer together, the fret effect is strengthened. As the protein stretches, moving the tags further apart, the fret effect weakens.
"By performing fret measurements together with one or more of the single-molecule manipulation techniques, it will be possible to correlate local protein structural changes with macromolecular function," says Weiss.
One of the biggest obstacles towards achieving this goal is the limitations of current fluorescent-dye molecules, but help is on the way. Recently Weiss and msd chemist Paul Alivisatos announced the development of a new type of fluorescent probe made from nanometer-sized crystals of semiconductors. These semiconductor nanocrystals offer a distinct advantage over conventional dye molecules in that they emit multiple colors of light, which means they can be used to label and measure several biological markers simultaneously.
"The use of semiconductor nanocrystals should allow us to do unique fret experiments," says Weiss. "For example, labeled molecules could be made to emit different colors at different times of an event."
Alivisatos, an authority on the production by chemical means of semiconductor nanocrystals, says, "The development of semiconductor nanocrystals for biological labeling would give biologists an entire new class of fluorescent probes for which no small organic molecule equivalent exists."
In an experiment on mouse tissue cells called 3T3 fibroblasts, a core nanocrystal of cadmium selenide was enclosed within a shell of cadmium sulfide and this core-shell combo was then enclosed within a shell of silica for water solubility and biocompatibility.
Since earlier research by Alivisatos had shown that the color of light emitted by a semiconductor nanocrystal depends upon its size, the mouse cells were labeled with two different sized core-shells. The silica surfaces of the core-shells were modified to selectively control their placement within the cells. Smaller nanocrystal core-shells, which fluoresced green, were modified to penetrate the nucleus of each cell; the larger core-shells, which emitted red light, were modified so that they would attach themselves to actin filaments along the outer cell membrane.

|
 |
 |
||
| Fluorescence Resonance Energy Transfer or FRET spectroscopy features a donor and an acceptor dye molecule attached at different points along a sample. When the donor is photo-excited at longer distances from the acceptor, fluorescence comes exclusively from the donor. As the space between them contracts, fluorescence is increasingly transferred to the acceptor. | ||||
Confocal microscopy images showed that cell nuclei had been penetrated with the green probes and the actin fibers had been stained red. The green and red labels were clearly visible to the naked eye and could be photographed in true color with an ordinary camera. Berkeley Lab is unique in that it is home to world-class facilities in the four main technological areas required for solving protein structures-synchrotron-based x-ray crystallography, nmr spectroscopy, electron microscopy, and supercomputing. Through its close ties to the Berkeley campus of the University of California, it brings together some of the top researchers in the biophysical sciences. The Laboratory is therefore well positioned to help meet what has been called the grand challenge in biology, characterizing and understanding how a cell's protein machinery works.
As pbd director Graham Fleming has said, "Nothing that takes place inside a living cell is a random occurrence; everything is being run by those amazing protein machines." - end -
| < Research Review | Top ^ | Next > |